
| ATS > Software > Scientific Programming with Python > Simulating a Magnet |
|
|
|||
NumPy provides a number of tools useful for simulations and for working with simple data types.
The Ising ModelThe Ising model is a simple model of a ferromagnet, using a regular lattice of magnetic “spins” that influence each other to line up, fighting against entropy all the while. Magnetic SpinsMagnets are atomic lattices with unpaired electrons, producing a spin magnetic moment at each position of the lattice: ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↓ ↓ ↑ ↑ ↑ ↑ → ← ↑ ↑ ↓ ← ↑ → → ↑ ↑ ↑ ↑ ↑ ↑ ↓ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↓ ↓ ↓ ↑ ↑ ↓ ← ↑ ↓ ← → ↓ ← → ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ← ← ↑ ↑ ↑ ↑ ↑ ↑ ← ↓ → ↓ ← ↑ ← ↑ ← ↓ ↑ ↑ ↑ ← ↑ ↑ ↑ ↑ ↑ ↑ ↑ ← ← ← ↑ ↑ → → → → ↓ ← → ← ↑ ← ↓ → ↓ → ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ → → → ↑ ← ↑ ↓ ↓ → ↑ ← → ↑ ↓ Low Temperature Medium Temperature High Temperature
Above a critical temperature there is no longer a dominant magnetic direction, and the domain magnetizations cancel each other out. At infinite temperature the spins are completely random in their orientation even over short distances. We can describe the tendency of the spins to line up in terms of the (potential) energy of the spin interaction. Over the entire lattice of size N it is given by E = – Σi≠j Jij si·sj where Jij is a positive coupling constant that incorporates the magnetic moment. Then,
Complete spin alignment will have a large negative energy, while thermally randomized spins will have a higher energy near zero. To simplify the model we can:
The energy thereby becomes E = –J Σnn sisj And we can represent the atomic lattice by an array of ±1 values: 1 -1 -1 1 1 -1 1 1 1 -1 -1 -1 -1 1 -1 -1 -1 1 1 1 1 -1 1 1 -1 1 1 -1 -1 1 -1 -1 1 -1 1 1 -1 -1 1 1 -1 -1 -1 1 -1 1 1 1 1 -1 1 -1 -1 1 -1 1 1 1 1 1 1 -1 -1 -1 1 -1 -1 -1 -1 -1 -1 -1 1 -1 -1 -1 1 -1 -1 -1 -1 -1 1 1 -1 -1 -1 -1 -1 1 -1 -1 1 1 -1 -1 -1 1 -1 1 This is known as the Ising Model of magnets, and as we will see it retains their basic characteristics. When the spins substantially align, there will be a measureable magnetization: M = (1/N)∣Σ si∣
The magnetization is an example of an order parameter, which we can use to monitor the system order. SpynsNot surprisingly, we can implement an Ising system and calculate its magnetization using Python. We’ll use a two-dimensional lattice of size N = L × L: L = 10 NumPy can define arrays with preset values in a number of ways. For example, to create a system at zero temperature, all spins aligned, we use: import numpy as np ⇒ The first argument to The second argument sets the data type, in this case NumPy integers (otherwise it would produce a float). The magnetization will tell us the degree of order in the model: M = lambda : np.absolute(np.sum(spins))/N ⇒ 1.0 Perfectly ordered, by design! Warning: in Python 2.x, you’ll want to use The function To create a system at infinite temperature, with a random distribution of values, we can use a subset of NumPy tools, import numpy.random as npr ⇒ The first two arguments to The third argument to The result is a NumPy array of integers (0, 1), which allows broadcast calculations, here multiplication by 2 and then subtraction of 1 to produce the desired values of (–1, 1). The magnetization is close to zero, as it should be for a completely disordered state: print('M = ', M()) ⇒ M = 0.06 It will never be precisely zero because it’s defined here, essentially, as the standard deviation of the average spin, but the larger the lattice we choose, the closer to zero it will be: M = (1/N)∣Σ si∣~ (1/N)×N1/2 ~ 1/N1/2 So for Spyn StatisticsFor a given temperature, how can we put this system in an equilibrium state so that we can calculate its order parameter? First we better define what we mean by “equilibrium”. Statistical mechanics tells us that, for a system in equilibrium at a temperature T, representing a bath of thermal energy that randomizes the spins, the probability of a particular configuration of the spins is proportional to its Boltzmann factor: P({si}) ∝ exp(–E/kT) where the Boltzmann constant k “converts” temperature to a corresponding energy. Clearly the lowest energy state will have the highest probability. For the Ising Model: P({si}) ∝ exp(–(–J Σnn sisj)/kT) where β = J/kT varies between ∞ and 0 as the temperature T varies between 0 and ∞. In particular,
Now consider an individual spin si and its neighbors sinn; it has an energy relative to its neighbors of Ei = –(J /2) si Σnnsinn where the factor of 1/2 avoids double-counting the energy once we include the neighboring spins. It’s helpful to define a spin sum Si as: Si = siΣnnsinn Ei = –(J /2)Si The probability of a particular microstate si given neighbors sinn is then: P(si|sinn) ∝ exp(–Ei/kT) Distinct examples of the individual spin sum, energy, and probability are:
We can again see that:
In addition, note that flipping the central spin will always invert the signs of Si and Ei. Now suppose we were to flip the central spin and this lowered the energy, e.g. we went from the second (blue) configuration in the table above to the last (pink) configuration. The spin sum is higher and the probability is also higher. On the other hand, if flipping the central spin increased the energy, e.g. going from the last (pink) configuration to the second (blue) configuration, the spin sum will be lower and the probabilty will also be lower. When in equilibrium the spins will flip back and forth over time, driven by the random energy of the temperature bath. However, on average, the system retains its state, in particular its degree of order. This means that the probability P of a microstate times the rate of transition W out of that microstate must be the same as the reverse process, i.e. over time the transition probabilities are equal: P(si|sinn)W(si → –si) = P(–si|sinn)W(–si → si) This condition is known as detailed balance, and is what we mean when we say a system is in equilibrium. Detailed balance can also be used to drive a nonequilbrium system to equilibrium by flipping spins in a compatible manner, and this is the key to simulating such a system. The ratio of the two transition rates is given by W(–si → si)/ W(si → –si) = P(si|sinn)/P(–si|sinn) Suppose flipping the spin si → –si lowers the energy; again this means that initially Ei > 0 and Si < 0. Because only the above ratio is important, we can choose this transition rate equal to 1, i.e. the spin will always flip: W(Ehigh → Elow) = 1 Consequently, when flipping the spin si → –si increases the energy, initially Ei < 0 and Si > 0, we must have W(Elow → Ehigh) = exp(–βSi) < 1 and the spin only has a fractional probability of flipping. Increasing the energy is therefore less likely than decreasing it, and after repeated flipping attempts the lower energy state will become the most probable state, so that a nonequilbrium system must approach equilibrium over time. Monte Carlo SimulationsBecause of the randomizing effect of thermal energy and the probabilistic nature of the system energetics, we can use random numbers to equilibrate a system. The Monte Carlo ProcedureSuppose we start with a system of spins at infinite temperature, which means they are randomly either ±1. We then instantaneously lower the temperature, a process known as quenching, and allow the system to equilibrate, or anneal. Alternatively, for low temperatures, we can start with a system at zero temperature, with a uniform distribution of either +1 or –1, and then instantantaneously raise the temperature and allow it to equilibrate. We can simulate such systems as follows:
This Monte Carlo procedure is a standard practice in statistical science as a way to calculate the properties of complex systems. It is sometimes called Markov-Chain Monte Carlo (MCMC), since each state in the calculation depends only the immediately previous state. It is also often referred to as the Metropolis Algorithm, after the first author of the 1953 paper Equation of State Calculations by Fast Computing Machines (who were listed in alphabetical order). However, the second author, computational physicist Arianna Rosenbluth, is credited with the actual implementation of the algorithm on one of the room-sized computers of the time, the MANIAC I at Los Alamos Scientific Laboratory in New Mexico:
The Monte Python SimulationWe’ll save the previous code in a file named import numpy as np We’ll also set a relatively large value of β that should produce some amount of ordering since it corresponds to a low temperature: beta = 2 Now the next steps will take place in a loop after generating the spins, starting by randomly picking an initial spin, as sequential picking of spins would generate spatial correlations: i = 0 This last line returns two values in the range (0, L–1) as a NumPy array. It is converted to a tuple 1 -1 -1 sl s sr 1 sb 1 -1 -1 -1 -1 1 sb -1 -1 1 1 1 1 -1 1 1 -1 1 1 -1 -1 1 -1 -1 1 -1 1 1 -1 -1 1 st sr -1 -1 1 1 st 1 1 sl s 1 -1 -1 -1 sl s sr 1 1 sb st -1 -1 1 -1 sb -1 -1 -1 -1 s sr 1 -1 -1 -1 1 -1 -1 sl sb -1 1 1 -1 -1 -1 st -1 1 -1 -1 1 1 st -1 sl s sr 1 Now locate the indices of the adjacent spins, left, right, bottom, and top: (sx, sy) = s sl = (sx-1, sy) sr = ((sx+1)%L, sy) sb = (sx, sy-1) st = (sx, (sy+1)%L) When the spin On the other hand, when the spin These are known as circular boundary conditions and they help to reduce errors associated with finite lattice size. Now calculate the spin sum Si = siΣnnsinn: S = spins[s] * ( spins[sl] + spins[sr] + spins[sb] + spins[st] ) Finally test for the energy condition: if S <= 0 : There is another random number generator here, Once i reaches N, this loop terminates. We can calculate the matrix and magnetization at this time to see how they have changed, with the following statements that are not indented so they are only calculated once: print(spins) Passing through the lattice once, on average, represents a single unit of time, since the entire lattice should have the chance to update once, no matter what its size. To run the loop for multiple units of time, define a number of time steps tmax = 100 EnsemblesThe calculations above are for particular lattice sizes, and over particular periods of time. But they are also for particular configurations of spins, which have a probability P({si}) ∝ Πiexp(–Ei/kT) = exp(–ΣiEi/kT) = exp(–E/kT) To calculate the order parameter M, we would use a weighted average over all configuration of the spins, known as an ensemble: ⟨M⟩ = Σi PiMi Since a large lattice will have a huge number of configurations, 2N, it’s impractical to calculate every one. However, a random selection of spins such as above can be repeated multiple times, and that should sample the configuration space. Each of the selected configurations will occur with its own relative probabilty, and after Z samples the average result should provide a good approximation, known as the sample mean: ⟨M⟩ = (1/(Z−1))ΣZ M (The factor of Z We can also calculate the sample mean of the magnetization squared: ⟨M2⟩ = (1/(Z−1))ΣZ M 2 which allows the calculation of the sample variance of M around the mean, given by: sM2 = (1/(Z−1))ΣZ(M − ⟨M⟩)2 and the sample standard deviation of M around the mean, given by: sM = sqrt(⟨M2⟩ – ⟨M⟩2(Z−2)/(Z−1)) The standard error of the mean of M then follows according to the Central Limit Theorem and is given by: σM = sM / sqrt(Z−1) All together, the code adds an extra loop for Z, with initializations beforehand, accumulations after each step, and final calculations afterward:
Z = 100
tmax = 100
m = 0 # to accumulate the magnetization
m2 = 0 # to accumulate the magnetization squared
Ms = () # to hold the individual magnetizations to display a histogram
z = 0
while z < Z:
z += 1
i = 0;
while i < tmax * N:
i += 1
s = tuple(npr.randint(0, L, 2))
(sx, sy) = s
sl = (sx-1, sy)
sr = ((sx+1)%L, sy)
sb = (sx, sy-1)
st = (sx, (sy+1)%L)
S = spins[s] * ( spins[sl] + spins[sr] + spins[sb] + spins[st] )
if S <= 0 :
spins[s] *= -1
else:
W = np.exp(-beta*S)
R = npr.rand()
if R < W : spins[s] *= -1
mi = M()
Ms += (mi,)
m += mi
m2 += mi**2
Mavg = m / (Z-1) # Average magnetization across the ensemble
M2avg = m2 / (Z-1) # Average magnetization squared across the ensemble
sM = (M2avg - Mavg**2 * (Z-2)/(Z-1))**0.5 # Sample standard deviation of M around the mean
sigmaM = sM/(Z-1)**0.5 # Standard error of the mean of M
Accumulating the magnetizations in a tuple
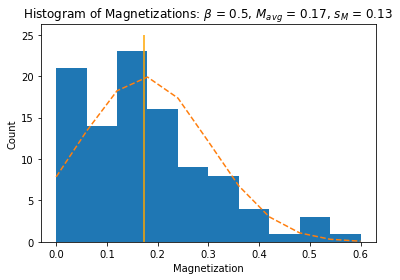
import matplotlib.pyplot as plt
figure, axes = plt.subplots()
n, bins, patches = axes.hist(Ms, 10) # the histogram of the data in 10 bins
# add a 'normal' fit curve
y = ((1 / (np.sqrt(2 * np.pi) * sM)) *
np.exp(-0.5 * (1 / sM * (bins-Mavg))**2))
y *= Z/np.sum(y) # scale to match total
axes.plot(bins, y, '--', color='orange')
axes.vlines(Mavg, 0, 25, color='orange')
axes.set_xlabel('Magnetization')
axes.set_ylabel('Count')
axes.set_title(r'Histogram of Magnetizations: $\beta$ = ' + str(beta) + \
', $M_{avg}$ = ' + str(round(Mavg,2)) + ', $s_M$ = ' + str(round(sM,2)))
plt.show()
plt.close()
High-Pyrformance ComputingSimulating a large number of variations of a physical model can take a lot of CPU time, and high-performance computing systems provide a lot of CPUs. The Amherst College High-Performance Computing System FrostByteLarger lattices can take a long time to process in the loop above, and they must then be averaged over a number of different configurations. While the script above should run well in Spyder, it will be very slow overall. We can therefore turn to the Amherst College High-Performance Computing System, named “FrostByte” by popular acclaim, which provides a CPU cluster with 1024 CPU cores on 16 Unix computers, and has Python already installed. On the CPU cluster we can run a number of calculations, or jobs, on these multiple cores and for longer periods of time. FrostByte jobs are prepared, tested, and submitted on an entry computer or login node, which you can access over the Internet from on-campus or while using a virtual private network (VPN) to appear to be on-campus. The FrostByte’s login nodes are accessed via the domain name General instructions for using the HPC system can be viewed in a web browser at https://www.amherst.edu/offices/it/academic-technology-services/tools/analysis-and-programming/high-performance-computing-system/frostbyte_documentation. Procedure 1: Connecting to the HPC SystemFrostByte’s computing environment requires the use of text commands rather than a graphical user interface, so you’ll need to open a command-line interpreter, also called a “terminal” or “shell” program, and log in to the login node of FrostByte over the network.
mkdir ising cd ising Procedure 2: Sharing Files with the HPC SystemTo submit jobs to the HPC system, you must copy your Python script over to it. You can use file sharing, SCP, or SFTP. File sharing will probably be more familiar to you, as the network drives appear as if they are local, like your G: drive.
Python Scripts on the HPC SystemOnce you’ve mapped a network drive to your local computer as in Procedure 2, you can also use the Spyder IDE to read and write files on it. Procedure 3: Setting Up Spyder to Use the HPC SystemTo work with your Python script on the HPC system, it’s simplest to set up Spyder to reference your working folder in
The To verify that the directory change worked, type IPython’s built-in “print working directory” command: %pwd ⇒ '/Volumes/username/ising' When running your script on the HPC system, there are a few changes you’ll want to make so that it works more simply with Unix and the HPC system’s job management software. Procedure 4: Making a Python Script Self-ExecutableTo simplify the execution of your script, you can make it run as a “self-executable” Unix application, rather than as a file that’s an argument for the Python interpreter.
#! /usr/bin/env python3 The first two characters are known as a shebang, and tell Unix to run the script as the next argument of the command that follows. chmod +x ising.py ls -l The three groups of permissions here, ./ising.py One additional useful capability is to be able to hand your executable one or more arguments that could have different values for different jobs in the HPC system. In Unix, such command-line arguments are typed following the initial command and separated by spaces, e.g. ./ising.py 1 where If you import the “system” or import sys then you will have access to command-line arguments, passed to your executable as a list of strings: sys.argv ⇒ ['./ising.py', '1'] beta = float(sys.argv[1]) ⇒ 1.0 Note that item In general you will want to test to make sure that the expected number of arguments are provided, and either set a default value for missing arguments or exit the program with an error message (another function provided by the if len(sys.argv) < 2 : Then when running the executable without arguments: ./ising.py ⇒ Usage: ./ising.py beta If you need to engage in more complicated argument-list processing, especially if you want to include options like One other important change for your executable is to format its output to make it easier to analyze large quantities of data; most simply, you can write comma-separated values so that the results of different jobs can all be concatenated together into a table: print('L,Z,beta,Mavg,sM,sigmaM') ⇒ The Slurm Job-Management SystemThe Slurm job-management software provides an efficient means to control a project running on a large number of computers. A set of related jobs are submitted as a cluster of jobs that are automatically distributed over any available computer cores. By design CPU time is shared with other researchers; the general behavior is that newer clusters are given priority, and the longer they run the lower their priority becomes. Along with your executable program, Slurm will need a batch file for each job cluster, which provides details on how to run each job, including any command-line arguments that might vary from job to job. The basic structure of a batch file is a shell script that provides instructions on the code to run along with a set of sbattch one per line, usually key=value pairs that describe either global job properties or individual job properties, for example:
#! /bin/sh
#SBATCH --ntasks=21
#SBATCH --output ising-%j.csv
#SBATCH --error ising-%j.err
# Check for the results directory, and if it isn't present, create it:
if [[ ! -d 'results' ]]; then mkdir 'results'; fi
for i in {0..20}
do
# Calculate the actual parameters using the index i, by running python inside ` `:
s=`python -c "print($i*0.1)"` # s in [0,2] in steps of 0.2
srun --overlap -N 1 -n 1 -o results/ising-%j-$i.out -e results/ising-%j-$i.err ./ising.py $s &
done
wait
# -n numeric sort
# -u unique lines (only one header)
# -r reverse sort (puts header at beginning)
sort -ur results/*.out
As with Python, a
Procedure 6: Running a Project on the HPC SystemOnce you have an application and a Slurm batch file describing how to run it, such as described above, there are just a few commands to know to get it running on the HPC system:
sbatch ising.sb ⇒ Submitting job(s) Note the job ID squeue --me ⇒ -- Submitter: csc2-node01.amherst.edu : <148.85.78.21:46911> ID OWNER SUBMITTED RUN_TIME ST PRI SIZE CMD 149415.0 username 12/18 16:56 0+00:03:34 R 5 9.8 ising.py 0.1 149415.1 username 12/18 16:56 0+00:03:34 R 5 9.8 ising.py 0.2 149415.14 username 12/18 16:56 0+00:03:34 R 5 9.8 ising.py 1.4 149415.15 username 12/18 16:56 0+00:03:34 R 5 9.8 ising.py 1.5 Total for query: 15 jobs; 0 completed, 0 removed, 0 idle, 15 running, 0 held, 0 suspended Total for username: 15 jobs; 0 completed, 0 removed, 0 idle, 15 running, 0 held, 0 suspended Total for all users: 15 jobs; 0 completed, 0 removed, 0 idle, 15 running, 0 held, 0 suspended With the option The actual command that Slurm is running is shown at the end, and you should verify that it’s what you expect. The status of each job is listed in the scancel 149415 149416.9 The command arguments are a list of cluster IDs and/or cluster.process IDs, or your user ID to remove all of your jobs. There are many more commands that might occasionally be useful and which are described in the Slurm documentation.
Procedure 7: Processing Your ResultsThe results of your cluster job are now stored in the directories
cat results/*/out > ising.csv The wild card character cat ising.csv ⇒ L,Z,beta,Mavg,sM,sigmaM 10,100,0.10,0.095,0.075,0.008 L,Z,beta,Mavg,sM,sigmaM 10,100,0.20,0.090,0.066,0.007 .... L,Z,beta,Mavg,sM,sigmaM 10,100,1.40,1.001,0.017,0.002 L,Z,beta,Mavg,sM,sigmaM 10,100,1.50,1.005,0.014,0.001 cat results/*.out | sed -e 1n -e /L/d > ising.csv The pipe character cat ising.csv ⇒ L,Z,beta,Mavg,sM,sigmaM 10,100,0.10,0.085,0.075,0.008 10,100,0.20,0.090,0.066,0.007 .... 10,100,1.40,1.001,0.017,0.002 Unix has a wealth of such commands to process the content of files, you can find a list of them on Wikipedia.
ising = np.loadtxt('ising.csv', delimiter=',', skiprows = 1)
ising
⇒
array([[1.000e+01, 1.000e+02, 1.000e-01, 8.500e-02, 7.500e-02, 8.000e-03],
[1.000e+01, 1.000e+02, 2.000e-01, 9.000e-02, 6.600e-02, 7.000e-03],
....
[1.000e+01, 1.000e+02, 1.400e+00, 1.001e+00, 1.700e-02, 2.000e-03],
[1.000e+01, 1.000e+02, 1.500e+00, 1.005e+00, 1.400e-02, 1.000e-03]])
import matplotlib.pyplot as plt
plt.plot(ising[:,2], ising[:,3])
plt.errorbar(ising[:,2], ising[:,3], ising[:,4])
plt.xlabel('β')
plt.ylabel('M')
plt.title(r'Ising Magnetization: $L$ = ' + str(L) + ', $Z$ = ' + str(Z))
plt.show()
plt.close()
Example: Running Jobs with Self-Generated DataSome jobs will generate all of their own data, typically from some statistical distribution, and so don’t need inputs via the command line. They do need to write their outputs in different files, and Slurm provides a means for that. Consider the following batch file “sample.sb”, which you can download here: #! /bin/sh #SBATCH --ntasks=50 #SBATCH --output=ising-%j.out #SBATCH --error=ising-%j.err # Check for the results directory, and if it isn't present, create it: if [[ ! -d 'results' ]]; then mkdir 'results'; fi # Hand the python script to Slurm to run, each task on its own cpu, and wait for them to complete: srun --overlap -o results/ising-%j-%2t.out -e results/ising-%j-%2t.err ./ising.py wait This is written for an R script, rather than Python, but hopefully the procedure is still clear.
ising = np.loadtxt('ising.out', delimiter=',')
ising
import matplotlib.pyplot as plt
plt.plot(ising[:,2], ising[:,3])
plt.errorbar(ising[:,2], ising[:,3], ising[:,4])
plt.xlabel('β')
plt.ylabel('M')
plt.title(r'Ising Magnetization: $L$ = ' + str(L) + ', $Z$ = ' + str(Z))
plt.show()
plt.close()
Problem 6: The Full MonteUsing the HPC system, repeat Procedure 7 over larger arrays L and more ensembles (e.g. Z = 1000), by generalizing the command-line input of your executable to allow run-time choices of these values. Calculate in each case the magnetization (using the approximation for large Z): ⟨M⟩ = (1/Z) ΣZ M the average square magnetization: ⟨M2⟩ = (1/Z) ΣZ M 2 and then the standard deviation of the mean value of M: sM = sqrt(⟨M2⟩ – ⟨M⟩2) Plot the results for different values of L on one graph; what happens as L gets larger?
|
||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||



 And then you can plot this data, this time adding error bars:
And then you can plot this data, this time adding error bars: